Visualizing BAM files hosted on s3
While the end product of many bioinformatics analyses are tables and graphs being able to visually inspect the alignment of sequencing data is valuable during method development or to diagnose issues with the sequencing library. Two excellent tools to visualize sequencing data are IGV and JBrowse 2. When running these tools locally sequence alignments stored in BAM files can be readily visualized. However, because sequencing data is large, and it is often stored on a remote cloud file system. While this is practical, the inconvenience of downloading alignment files and installing visualization software can be so great that opportunities to make insights through data visualization can be easily missed.
Luckily, when hosting sequence alignments on S3 it is easy to make this data more accessible to users in your organization through their web browser.
Accessing Data via HTTP
While objects stored on S3 are typically
referred to using their S3 URI ( e.g.,
s3://example/file.bam ), every s3 resource has
can also be referred to by an HTTP URI. For
example, a file stored on S3 at location
s3://example/file.bam can be retrieved by HTTP
using its
Object URL
https://s3.amazonaws.com/example/file.bam, and
if bucket permissions allow public access the
files can be readily downloaded using a web
browser or command line tools like
curl
and
wget. However, objects stored in an s3 bucket are
private by default. Private objects can also be
retrieved using HTTP using
presigned URLs. Since the process of loading data in IGV via
HTTP is the same using both signed and unsigned
object URLs my example below uses objects stored
in a publicly accessible bucket for simplicity.
CORS headers
In order for the IGV genome browser application the runs in the user’s browser to be able to retrieve sequencing data from S3 the server where the data is hosted has to be configured to allow “cross-origin” requests. By default web browsers prevent a web page from displaying content from different hosts. The Cross-origin resource sharing (CORS) protocols enables servers to allow exceptions to this default behavior to be made. In the case of igv.js we will use CORS to allow the JavaScript genome browser application to be hosted independently of the data that is displayed.
The igv.js wiki provides guidance on how the S3 bucket CORS headers can be configured to make them accessible to igv.js.
Basic IGV.js configuration
The configuration of igv.js is described in the Quickstart guide. A minimal setup to embed IGV in a webpage for a genome browser session with data assets hosted off of s3 would be
index.html:
<html>
<header>
<!--1-->
<script src="https://cdn.jsdelivr.net/npm/igv@2.15.8/dist/igv.min.js"></script>
</header>
<body>
<h1>IGV</h1>
<!--2-->
<div id="igv-browser"/>
<script>
<!--3-->
var myTrack = {
"name": "my track",
"url": "https://s3.amazonaws.com/1000genomes/file.bam",
"indexURL": "https://s3.amazonaws.com/1000genomes/file.bam.bai",
"format": "bam"
}
var options = {
genome: "hg38",
locus: "chr1:1-100",
tracks: [ myTrack ]
};
<!--4-->
var igvDiv = document.getElementById("igv-browser");
igv.createBrowser(igvDiv, options)
</script>
</body>
</html>
To walk through this file:
-
The source for igv.js is loaded from the developer-hosted content deliver network distribution for igv.js
-
A placeholder in the markup is created so that the IGV browser can be injected into the page (see step 4)
-
IGV is configured to specify the reference genome and tracks to be displayed
-
The browser is initialized and injected into the page
Combining Shiny with IGV-js
While configuring a genome browser to visualize data is a good first step, serving data off of S3 can really shine when a the browser is is embedded as a component in a data-rich web-application. To illustrate how genomics data hosted on s3 can be visualized I will create a small Shiny app that allows a user to select a dataset to visualize by selecting from a list:
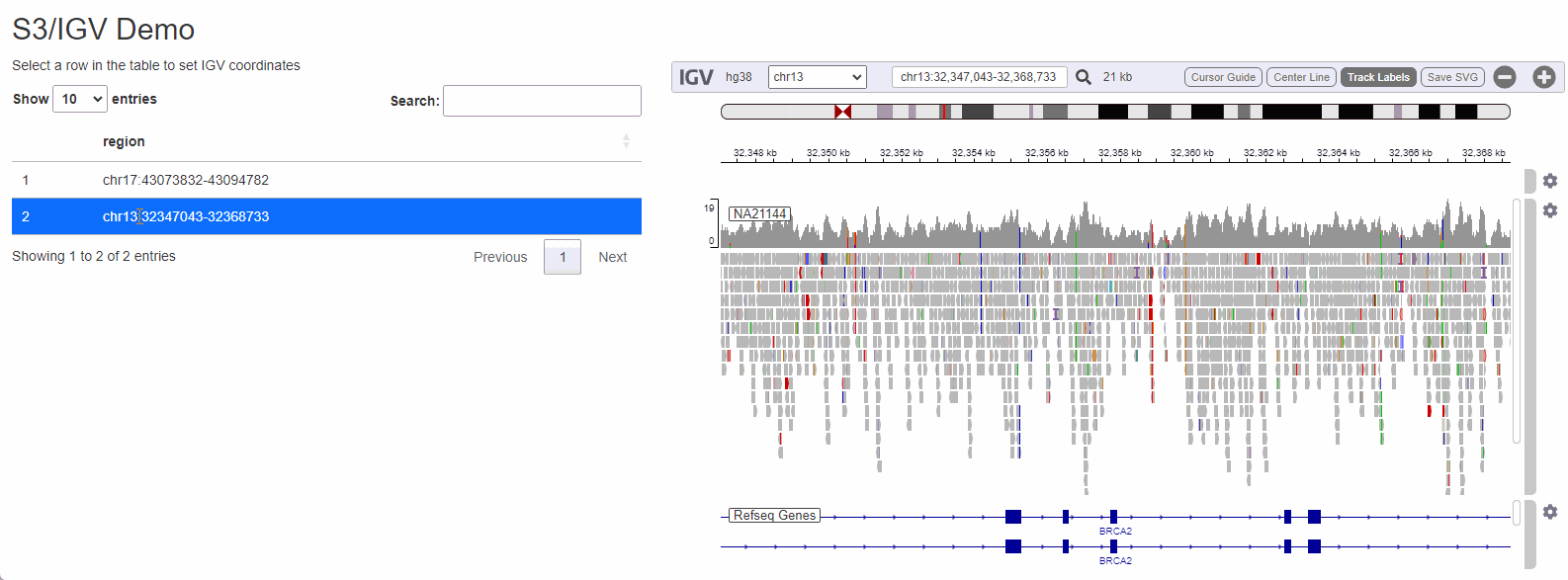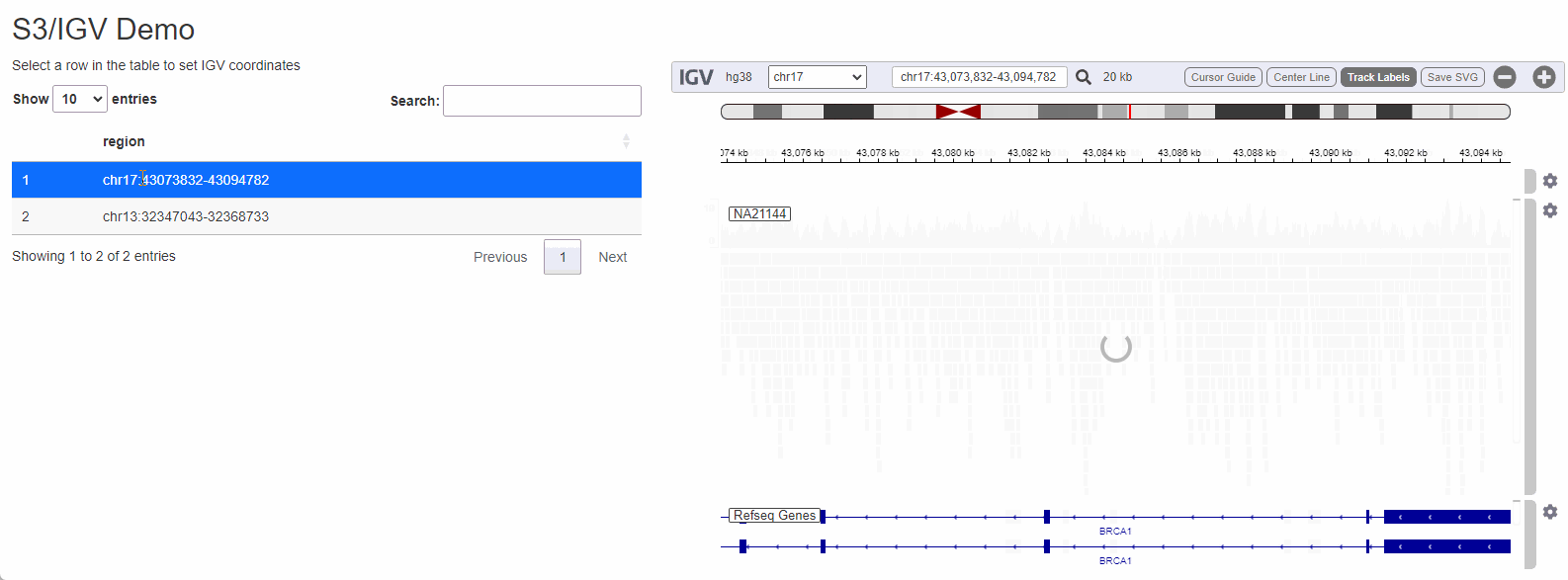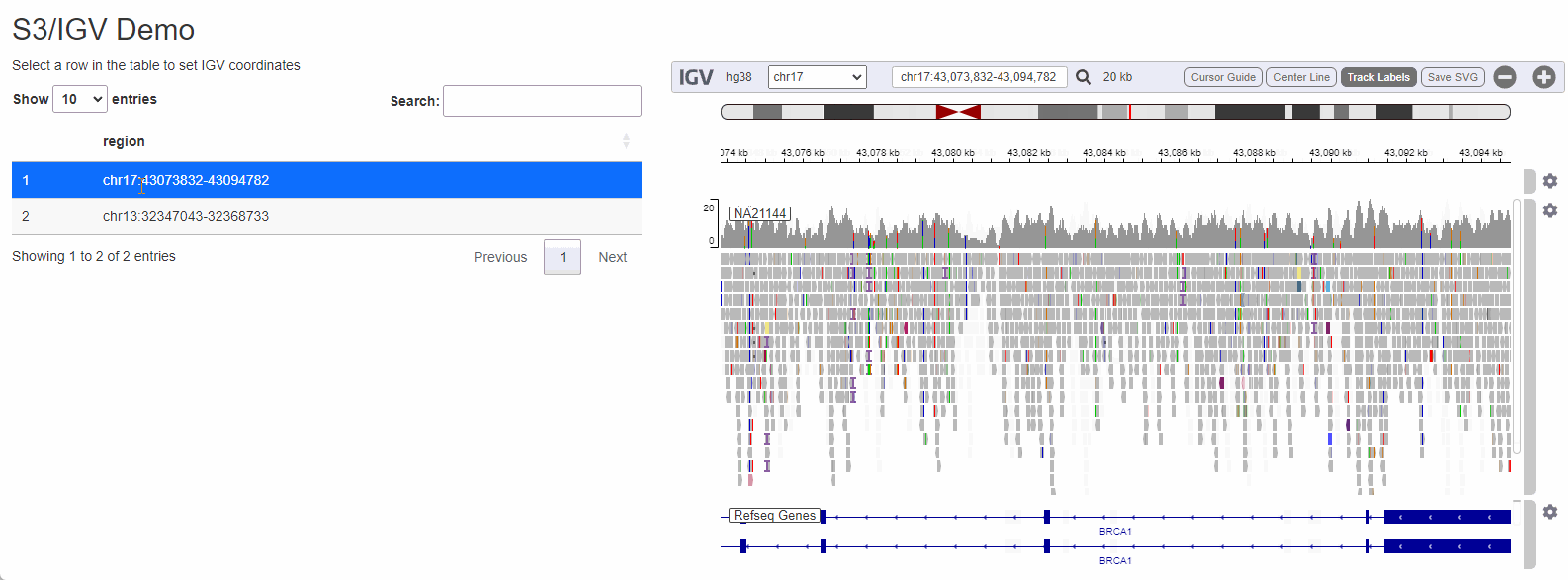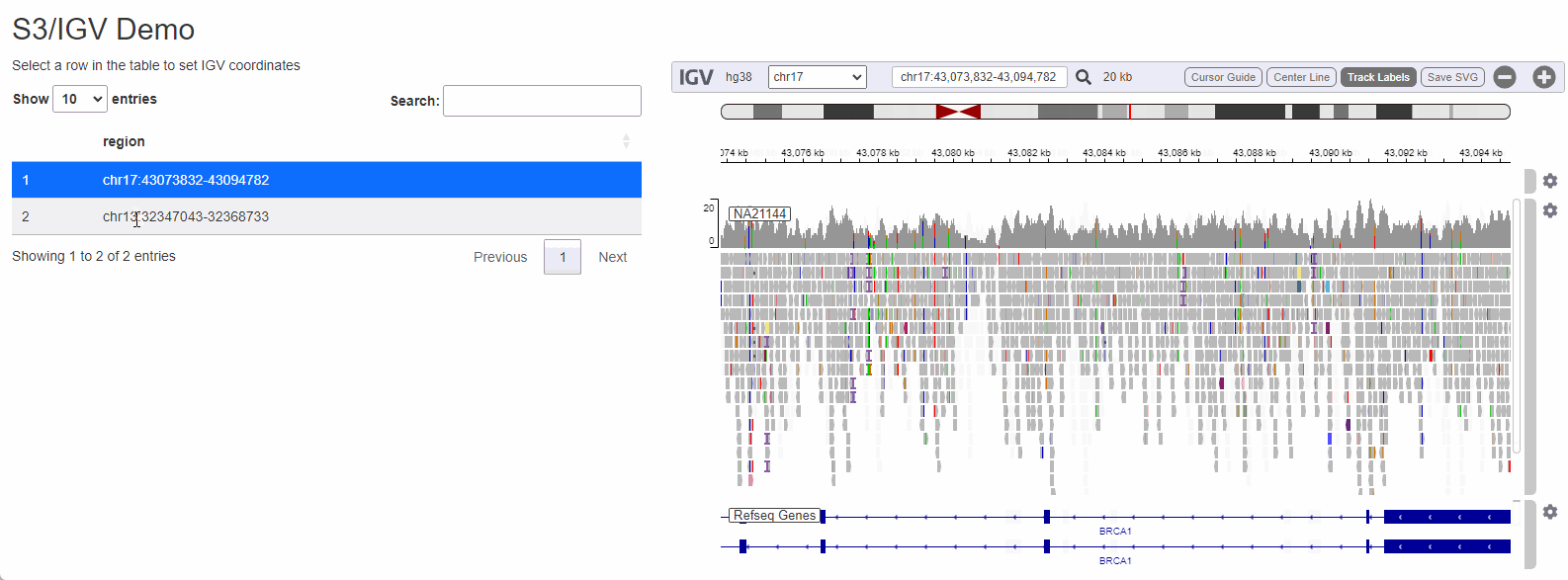
Shiny is a framework for R to makes it easy to build web applications that provide a no-code interface for people to interact with data and take advantage of the language’s powerful statistics/machine learning/data visualization capabilities. Being able to cross-reference raw sequencing data from a Shiny application can streamline a process that would otherwise be cumbersome (locating the BAM file corresponding to the sample, copying it from remote storage to the local computer, opening IGV and configuring it to visualize the region of interest).
The process to wire IGV-js into a Shiny app is
analogous to the approach used in the first
example above: In the UI block the IGV-js
library is loaded, a
<div>
is created for the browser, and using a
script
tag specify that the browser should be created
when the page loads. In the
server
function a table of genomic regions is created
with an observer so that when a row of the table
is selected the IGV-js API is used to set the
coordinate of the genome browser that is
displayed.
To provide a high level overview, the app is composed of an R script and a little JavaScript:
├── app.R
└── www
└── setup_igv.js
With UI and server components defined:
# file: 'app.R'
library(shiny)
ui <- fluidPage(
tags$head(
tags$script(src='setup_igv.js'),
tags$script(src='https://cdn.jsdelivr.net/npm/igv@2.15.8/dist/igv.min.js')
),
titlePanel("S3/IGV Demo"),
fluidRow(
column(5,
tags$p('Select a row in the table to set IGV coordinates'),
DT::DTOutput('regions')),
column(7,
div(id='igv-browser'))
)
)
server <- function(input, output, session) {
regions_table <- tibble::tibble( region = c( 'chr17:43073832-43094782', 'chr13:32347043-32368733') )
output$regions <- DT::renderDT( DT::datatable( regions_table, selection = 'single' ) )
observeEvent( input$regions_rows_selected,{
selected_region <- regions_table[input$regions_rows_selected,]$region
session$sendCustomMessage('igv-search', selected_region)
})
}
shinyApp(ui, server)
Where the snippet used to configure the IGV browser is:
// file: 'www/setup_igv.js'
document.addEventListener("DOMContentLoaded",function(){
var s3_track = {
name: "NA21144",
url: "https://s3.amazonaws.com/1000genomes/data/NA21144/alignment/NA21144.alt_bwamem_GRCh38DH.20150718.GIH.low_coverage.cram",
indexURL: "https://s3.amazonaws.com/1000genomes/data/NA21144/alignment/NA21144.alt_bwamem_GRCh38DH.20150718.GIH.low_coverage.cram.crai",
format: "cram"
}
var options = {
genome: "hg38",
tracks: [s3_track]
};
var igvDiv = document.getElementById("igv-browser");
browser = igv.createBrowser(igvDiv, options);
Shiny.addCustomMessageHandler('igv-search',function(txt){
browser.then(function(igvb){ igvb.search(txt) });
});
})
And the app can be run by calling:
Rscript -e 'shiny::runApp(".")'
Shiny / IGV-js communication
In order to use the IGV-js library from Shiny
the Shiny UI needs to be able to control the
behavior of the IGV browser. IGV-js provides an
API
to drive browser functions like setting the
genome version, navigation, configuring tracks.
To call these API methods we need to be able to
“send messages” from the Shiny framework to
JavaScript running on the client’s web browser.
Shiny provides a mechanism to do this via the
session$sendCustomMessage()
function. By defining a new event type
‘igv-search’ we can send a request from Shiny to
javascript running on the client with the
coordinates of the region to be displayed:
session$sendCustomMessage('igv-search', selected_region)
This message is received by a message handler that listens for events with the specified type, and calls the appropriate IGV API method when the event occurs.
Shiny.addCustomMessageHandler('igv-search',function(txt){
browser.then(function(igvb){ igvb.search(txt) });
});
Summary
Visualizing data in a genome browser can provide unique insights that are otherwise obscured by high-level summary statistics. Saying “it’s important to visualize your data” isn’t enough, put visualizations so front-and-center that the team has no choice but to ogle it a little as they review the higher-level analysis. Doing so will encourage serendipitous discoveries, and help to avoid being misled by summary statistics that mask weird edge cases.
If you are storing your sequencing data on S3 this pattern is great because you can build rich data-applications without any added cost – no additional server needs to be running 24/7 just to serve the files.
References
James T Robinson, Helga Thorvaldsdottir, Douglass Turner, Jill P Mesirov, igv.js: an embeddable JavaScript implementation of the Integrative Genomics Viewer (IGV), Bioinformatics, Volume 39, Issue 1, January 2023, btac830, https://doi.org/10.1093/bioinformatics/btac830
Comments